
Pieter Tans and Kirk Thoning, NOAA Global Monitoring Laboratory,
Boulder, Colorado
September, 2008. Updated December, 2016; March 2018, September 2020
Note: This is an update that incorporates new measurement methods and analyzer at Mauna Loa. The previous version of this document that discusses the infrared analyzer measurments at Mauna Loa is available here.
We have confidence that the CO2 measurements made at the Mauna Loa Observatory reflect truth about our global atmosphere. The main reasons for that confidence are:
Most people assume that we measure the “concentration” of CO2 in air, and in communicating with the general public we frequently use that word because it is familiar. The quantity we actually determine is accurately described by the chemical term “mole fraction”, defined as the number of carbon dioxide molecules in a given number of molecules of air, after removal of water vapor. For example, 413 parts per million of CO2 (abbreviated as ppm) means that in every million molecules of (dry) air there are on average 413 CO2 molecules. The table below gives approximate values of gases in the atmosphere for 413 ppm of CO2 in dry air (this is roughly the average amount of CO2 in the atmosphere in the middle of the year 2020). All species have been expressed as ppm, turning 78.09% nitrogen into 780,900 ppm. The rightmost column shows the composition of the same air after enough water vapor has been added to make the mole fraction of water vapor in wet air 3%:
| dry air | 3% wet air | |
|---|---|---|
| Nitrogen | 780,900 | 757,473 ppm |
| Oxygen | 209,360 | 203,079 |
| Water vapor | 0 | 30,000 |
| Argon | 9,300 | 9,021 |
| Carbon Dioxide | 413 | 400.6 |
| Neon | 18 | 17.5 |
| Helium | 5 | 4.9 |
| Methane | 2 | 2 |
| Krypton | 1 | 1 |
| trace species (each less than 1) | 1 | 1 |
| Total | 1,000,000 | 1,000,000 ppm |
The concentration of a gas is defined formally as the number of molecules per cubic meter. The goal of our measurements is to quantify how much CO2 has been added to, or removed from, the atmosphere. The concentration does not give us that information because it primarily depends on the pressure and temperature, and secondarily on how much the relative abundance of each gas has been diluted by water vapor, which is extremely variable. Only the dry mole fraction reflects the addition and removal of a gas species because its mole fraction in dry air does not change when the air expands upon heating or upon ascending to higher altitude where the pressure is lower. Nor does it change when water evaporates, or condenses into droplets. Why is this so important? Here is an example: The amount of CO2 is higher in the Northern than in the Southern Hemisphere as a result of the combustion of coal, oil, and natural gas. The measurement of this difference gives us crucial quantitative information about the emissions and removals of CO2. The concentration change produced by the addition of water vapor can be greater than the CO2 difference between the two hemispheres. In contrast, the difference in dry mole fraction does reflect the differences in emissions and removals between the hemispheres.
One often encounters in the literature the term “mixing ratio”. This is ambiguous. Does it apply to mass or to the numbers of molecules? In an attempt to clear that up one also often finds “mixing ratio by volume”, abbreviated as “ppmv” for CO2. However, that can still be ambiguous. Does it mean partial molar volume of CO2 relative to molar volume of whole air? That would only be correct if all the constituent gases of air behaved as ideal gases, which we know is not true. Furthermore, the calibration standards used for greenhouse gases in air are made as, and expressed as, mole fractions in (dry) air. All national metrology institutes, with NOAA/GML representing the WMO Global Atmosphere Watch community (see gml.noaa.gov/ccl/ ), participate in comparisons of reference gas standards that are called “Key Comparisons of Amount of Substance Ratios”. The unit of “amount of substance” is the mole.
The CO2 data that is described on this page is available at the GML Data Finder, where data options have been filtered to show CO2 data from all of the GML Observatories (In addition to Mauna Loa, Barrow Alaska, American Samoa and South Pole are included). Data is available as hourly, daily and monthly averages.
Although we describe measurement techniques for CO2, they apply also to measurements of methane (CH4) and carbon monoxide (CO).
In April of 2019, a new CO2 analyzer was installed at Mauna Loa that uses a technique called Cavity Ring-Down Spectroscopy (CRDS). (Prior to this date, an analyzer was used based on infrared absorption.) CRDS is based on the measurement of the rate of absorption, rather than the magnitude of absorption, of light circulating in an optical cavity. The beam from a laser enters an optical cavity consisting of two or more highly reflective mirrors. The laser beam is reflected back and forth inside the cavity, the so-called ‘ring-down cavity’. The laser is then turned off and the light intensity inside the cavity steadily leaks out and decays to zero in an exponential fashion. A detector measures the intensity of the transmitted light as a function of time. The decay time is called the cavity ring-down time. By comparing the ring down times when the laser is at a wavelength that the CO2 molecule does not absorb, to the ring down time when the laser is at a wavelength that the CO2 molecule does absorb, the amount of CO2 can be calculated (The analyzer also measures CH4 and CO).
An important aspect of the measurements is the ongoing calibrations of the analyzer. Air flows continually through the instrument, after having first been dried in a cold trap where the water vapor freezes out as ice on the walls of the trap. Unfortunately, the absorption that we measure in the instrument does not depend on the CO2 mole fraction, but on the total amount of CO2. Therefore, we also accurately control the temperature and pressure in the instrument, as well as the flow rate, and we do frequent calibrations with reference gas mixtures of known amounts of CO2-in-dry-air (stored in high pressure aluminum cylinders) spanning the expected range of the measurements.
We continuously record the output from the analyzer, but we need some way to convert this output to true mole fraction values, that is, M.F. = ƒ(ao); the mole fraction amount is some function of the analyzer output. The method we use to obtain this function is to use five “standard” gases of very well known values of CO2, with a wide range of values that span the entire range of natural ambient CO2 values. In the case of Mauna Loa, the range of the standards is from about 350 to 430 ppm. Each of the five standards is compared several times with a “Reference” gas from a tank of CO2-in-dry-air called “R0”, and the average difference between each standard and the reference gas is computed. We then plot up each of these differences (ΔR0) vs. the known mole fraction of CO2 in the standards, and fit a polynomial function that gives us
that is, the mole fraction amount of our sample is a function of the difference between the sample and the reference R0. c0, c1 and c2 are the coefficients of the polynomial function. This calibration sequence is performed approximately every two weeks. The calibration method allows us to determine the response of the analyzer without having to know the amount of CO2 in the reference R0, and the highly calibrated standard gases are used sparingly.
An example of the output from the analyzer for one day is shown in Figure 1. Two separate intake lines are used for sampling ambient air. The intake lines are from the top of a 38 m tall tower next to the observatory, to avoid any influence on the measurements by human activities at the observatory. Each intake line is measured for five minutes, alternating between line 1 and line 2. Once an hour, the reference gas R0 is measured for five minutes. Once per day, two target tanks are measured for fifteen measurements each (seen in hour 6 and hour 17 in Figure 1). The first few minutes after each gas change are not used to allow time for the previous gas to be completely flushed from the analysis system. The difference of the ambient air measurements from the reference R0 are calculated, and these differences are put into equation 1 to calculate the true ambient mole fraction CO2. By making the measurements relative to the difference from R0, any short term drifts in the analyzer are accounted for.
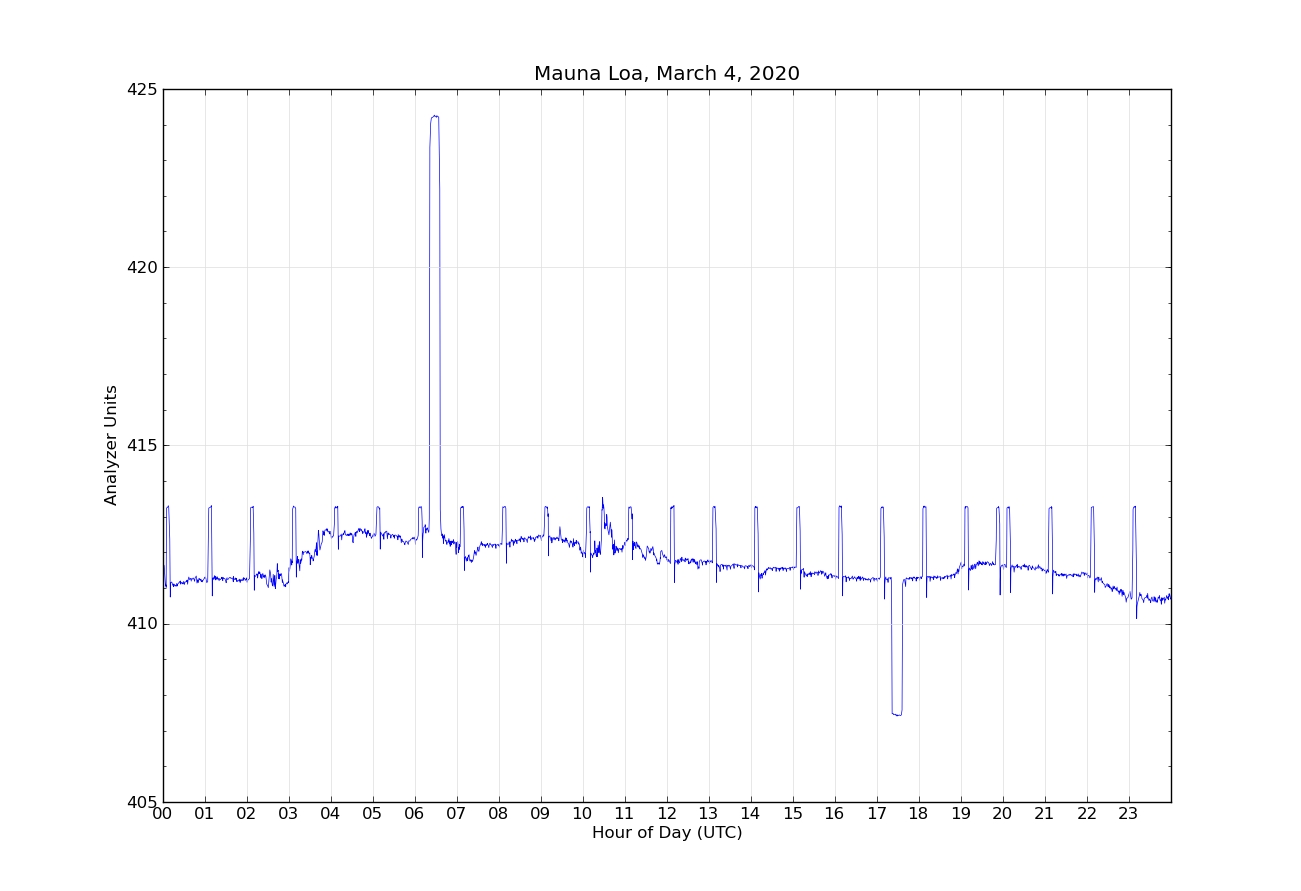
The calibration of the analyzer outlined above depends on accurate measurements of the standard gases. We provide for a check on the calibration response by running what we call two “target gases”, treated as having an unknown mole fraction, alternating every 11 hours through the measurement system. In reality the mole fraction of the target gas is very accurately known. If the response of the analyzer as calculated in Equation 1 is incorrect, the measurement of the target gas will produce the wrong result. By comparing the calculated values of the target gases with their known calibrated CO2 values, we can detect problems with the analyzer calibration. One target gas is near the ambient levels of CO2, and the other is higher than ambient. The repeat cycle of every 11 hours causes the target measurement to slowly move through the diurnal cycle because each day it is measured two hours earlier than on the previous day. This helps in determining if there are any problems related to the time of day.
In 1957 Dave Keeling, who was the first to make accurate measurements of CO2 in the atmosphere, chose the site high up on the slopes of the Mauna Loa volcano because he wanted to measure CO2 in air masses that would be representative of much of the Northern Hemisphere, and, hopefully, the globe. That goal has not changed. We still want to eliminate the influence of CO2 absorbed or emitted locally by plants and soils, or emitted locally by human activities. Dave Keeling also introduced the principle of a rigorous calibration strategy that we still employ today.
The observatory is surrounded by many miles of bare lava, without any vegetation or soil. This provides an opportunity to measure “background” air, also called “baseline”” air, which we define as having a CO2 mole fraction representative of an upwind fetch of hundreds of km. Nearby emission or removal of CO2 typically produces sharp fluctuations, in space and time, in mole fraction. These fluctuations get smoothed out with time and distance through turbulent mixing and wind shear. A distinguishing characteristic of background air is that CO2 changes only very gradually because the air has been mixed for days, without any significant additions or removals of CO2. Another common word for emissions is “sources”, and for removals, “sinks”.
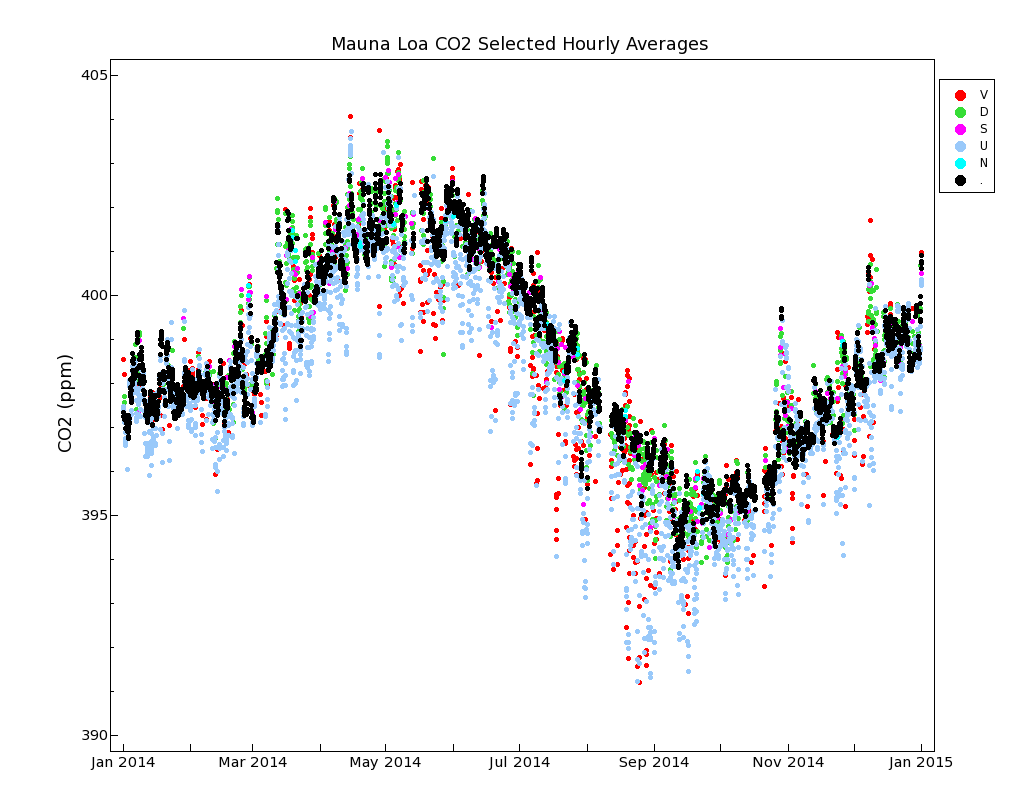
Figure 2 shows an example of the data selection procedures we use to categorize air that has likely been influenced significantly by nearby sources/sinks. The plot is for two days, July 18 and 19, 2020. First the time axis needs to be explained. The bottom axis of Figure 2 reflects that we keep date and time at the Mauna Loa Observatory in Universal Coordinated Time, abbreviated as UTC. UTC is the same across the entire world. It succeeded Greenwich Mean Time in 1961, and is widely used and unambiguous. Local time in Hawaii lags UTC by 10 hours, so that 10 am UTC corresponds to midnight locally. The upper axis has labels indicating the local time in Hawaii.

At Mauna Loa we use the following data selection criteria:
The standard deviation of the 5-minute mole fraction averages should be less than 0.30 ppm within a given hour. A standard deviation larger than 0.30 ppm is indicated by a “V” flag in the hourly data file, and by the red color in Figure 2.
There is often a diurnal wind flow pattern on Mauna Loa driven by warming of the surface during the day and cooling during the night. During the day warm air flows up the slope, typically reaching the observatory at 9 am local time (19 UTC) or later. The upslope air may have CO2 that has been lowered by plants removing CO2 through photosynthesis at lower elevations on the island, although the CO2 decrease arrives later than the change in wind direction, because the observatory is surrounded by miles of bare lava. Upslope winds can persist through ~7 pm local time (5 UTC, next day, or local hour 19 in Figure 2). Hours that are likely affected by local photosynthesis (11am to 7pm local time, 21 to 5 UTC) are indicated by a “U” flag in the hourly data file, and by the blue color in Figure 2.
In keeping with the requirement that CO2 in background air should be steady, we expect the hourly average from one hour to the next should not differ from the preceding hour by more than a small amount. Data where this hour-to-hour change exceeds 0.25 ppm is indicated by a 'D' flag in the hourly data file, and by the green color in Figure 2. However, if there are 3 consecutive non-flagged hours that either preceed or follow an hour with a 'D' flag, then the 'D' flag is removed. This handles a 'step' change in CO2 concentration, where the edges of the step agree with surrounding hours, but not with the single adjacent hour.
After the application of the 'V', 'U' and 'D' flags, there can be times when a single hour remains unflagged, but is bracketed by flagged data. This makes it unclear if this single hour could be representative of background air or not. We therefore apply a 'S' flag to these single hours. Hour 6 UTC on July 19 in Figure 2 is an example of this. Similarly, a pair of hourly data could be bracketed by flagged data. As a final step, we check if there are any unflagged data within ± 8 hours of an hourly average, ignoring adjacent hours. If not, then that hour is given a 'N' flag.
Hours for which we do not have valid air measurement are flagged with either a '*' or 'I' character in the hourly data file. The hours during which we do a standard gas calibration are an example, but there are also other instrumental causes, such as mechanical problems with pumps and valves.
The frequency for each of these flags for a typical year (2014) is shown in Table 1.
| Flag | Number of Hours | Percentage of year* |
|---|---|---|
| No flag | 3323 | 37.9% |
| V | 578 | 6.6% |
| U | 2680 | 30.6% |
| D | 1063 | 12.1% |
| S | 232 | 2.6% |
| N | 34 | 0.4% |
| * or I | 858 | 9.8% |
| *There are 8760 hours in the year. | ||
Hourly means are calculated wherever possible, and how we use that data is indicated by the selection flags. No data are thrown away. The hourly averages are calculated from the 'raw' data, which are the analyzer output values recorded for the air measurements as well as for the reference gas mixtures used for calibration and for the target gas.
On average over the entire record there are 9.0 retained hours per day with background CO2 mole fractions. Because of the upslope wind condition flag 'U', there is only a maximum of 15 hours per day of background values possible. Figure 3 show a plot of the CO2 data for 2014 for each flag type. Even though the data selection method identifies background data properly most of the time, there can still be conditions where the applied selection flag is not appropriate. The user of the data should be aware of this and modify the selection as needed.
System status variables measured and continuously recorded are: analyzer temperature, cold trap temperature, room temperature, sample flow rate, and both pressure and flow rate through both air intake lines from the tower.
Since 1995 our laboratory has been the CO2 Central Calibration Laboratory of the World Meteorological Organization (WMO). Before 1995 that role was filled by the Scripps Institution of Oceanography of the University of California in San Diego. We maintain the WMO Mole Fraction Scale for CO2-in-air. The WMO Scale is based on 15 so-called Primary Standards that are calibrated in terms of fundamental quantities at intervals of ~1.5 year by manometric measurements. They have CO2 mole fractions that span the range of ambient air. The primaries are stored in high pressure aluminum cylinders. The Primary Standards themselves are only sparingly used, so that we can build up a calibration history for each of them over several decades.
During a calibration episode, lasting 2-3 months, each Primary is measured three times. Each time a sample of air is taken from the cylinder and its pressure and temperature inside a closed volume are measured very accurately, after temperature and pressure have stabilized in a temperature controlled environment. The volume of approximately 6 liters is accurately known. Once the pressure, temperature, and volume of a gas are known, the amount of gas can be calculated, taking the temperature dependent compressibility of the gases into account. Then the air, with the CO2 that is to be determined still in it, is slowly and completely flowed at low pressure over a cold trap cooled with liquid nitrogen. The condensate in the cold trap consists of carbon dioxide, nitrous oxide, and residual water vapor. The latter is low, corresponding to 1 ppm or so in the dry high pressure cylinder. After separation of the water vapor component, the carbon dioxide and nitrous oxide are transferred to a small volume, about 10 cc, which has also been very accurately calibrated. After stabilization, the pressure and temperature are once again recorded, and total amount of the two remaining gases calculated. At that point we have determined the combined mole fraction of carbon dioxide and nitrous oxide in the cylinder air. The latter typically comprises less than one thousandth of the total, and we correct for its contribution after measuring nitrous oxide separately on a gas chromatograph.
The accuracy of the WMO Scale has been estimated, based on the accuracy of the temperature, volume and pressure measurement, and the gas handling procedures, at 0.10 ppm. It has been compared several times to calibration scales established independently by other laboratories that were based on weighing a small amount of pure carbon dioxide that was then mixed into a large, also accurately weighed, amount of carbon dioxide-free air. We have also compared to the previous WMO Scale maintained by Scripps. These comparisons are compatible with our internal accuracy estimate.
The manometric calibrations are a very time consuming process, while we need to calibrate thousands of cylinders, many of which are used by other laboratories around the world. We transfer, or propagate, the WMO scale to other high pressure cylinders of CO2-in-air in a much more efficient way, by comparing them to each other on an infrared gas analyzer system. This is very similar to how we measure outside air by comparing the voltage response of unknown air to the response of air with known mole fractions. The repeatability of these transfer calibrations, done one week or more apart, is typically 0.01-0.02 ppm in the range of ambient air. The sequence is similar to that of target gas calibrations at the field observatories where all cylinders are measured repeatedly, one after the other, with the known gases interspersed between the unknown gases. Because we want to use the Primary Standards sparingly, we transfer the scale twice per year through these comparisons to Secondary Standards, which in turn get used to calibrate all other reference gases. The Secondaries typically last for 3-5 years.
An important aspect of the calibration strategy is that reference gas mixtures are not only calibrated before use, but also after their use when the pressure is low, but not close to zero. Reference gas mixtures are recalled for recalibration when ~20% of the air is still in the cylinder because experience has shown that the CO2 mole fraction tends to drift more when the pressure becomes low. Because the raw data consists of voltages, it is possible to re-calculate corrected mole fractions for the measured atmospheric air after it has become clear through recalibration that a particular reference gas has drifted. Fortunately this problem greatly diminished when we made the switch from using steel cylinders to aluminum cylinders.
We know from experience that careful calibration is necessary, but not sufficient, for making accurate measurements. There are many potential biases related to gas handling, drying, that are not covered through the calibration. The ultimate test of how accurate these measurements are likely to be is to compare them with other measurements, done independently, with different methods, and/or by different laboratories. At Mauna Loa we have both. In Figure 4 we show a comparison with two sets of grab samples of air in glass flasks, done weekly in pairs. One set, called “S”, are samples taken through the intake line used by the continuous analyzer, and diverted just before entering the analyzer. The second set, called “P”, are completely independent from the continuous analyzer system. They are taken with a battery operated pump, and extendable air intake mast, at a spot on the lava away from the observatory buildings. The flask samples are sent to our laboratory in Boulder, Colorado, where they are analyzed together with thousands of other air samples from all over the world. There is a fair amount of “scatter” in the comparisons, partly because we are comparing a spot sample, a ~20 s average, to the longer average of the continuous analyzer when the sample was taken. On average, the “P” flasks differ from the continuous analyzer by 0.12 ppm, and the “S” flasks by 0.08 ppm. Important are systematic biases that persist over a good part of a year and more, portrayed by the solid smooth lines fitted to the respective data sets. One can see that most of the time the biases are less than 0.2 ppm.
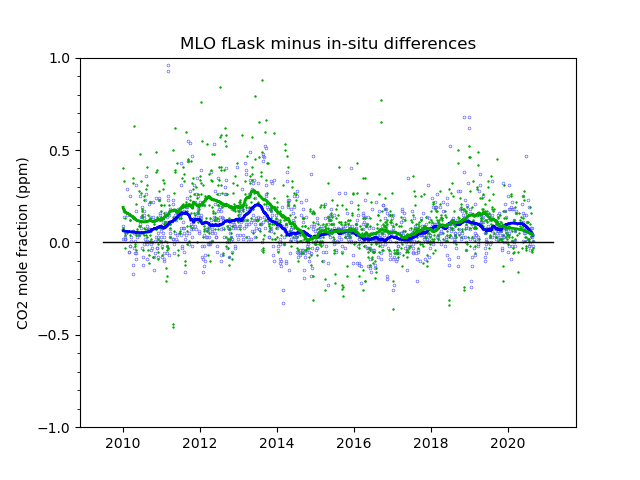 Figure 4. The figure shows differences between grab samples of air in glass flasks, and the corresponding hourly mean CO2 value from the Mauna Loa in-situ analyzer. There are two types of flask samples, the 'P' type collected using an independent flask sampling package (green pluses), and the 'S' type, collected using the in-situ analyzer air intake lines (blue circles).
Figure 4. The figure shows differences between grab samples of air in glass flasks, and the corresponding hourly mean CO2 value from the Mauna Loa in-situ analyzer. There are two types of flask samples, the 'P' type collected using an independent flask sampling package (green pluses), and the 'S' type, collected using the in-situ analyzer air intake lines (blue circles).
We have a similar comparison with the CO2 measurements performed by the Scripps Institution of Oceanography, the program started by David Keeling. They have always maintained their own manometric calibration scale, they use a different analyzer system, and their data selection techniques for selecting background air are different from, and independent of, our methods. They also compare on a regular basis with their own grab samples. A comparison of the Scripps monthly mean CO2 data (obtainable from https://scrippsco2.ucsd.edu/data/atmospheric_co2/) shows that the average difference during 1974-2019 between the Scripps and our monthly means is 0.07 ppm.
The above comparisons give us confidence that the CO2 measurements are generally accurate to better than 0.2 ppm.
We present two typical examples of measured CO2 mole fractions over the continent, to demonstrate what an excellent choice Mauna Loa is for making background measurements. Figure 5a shows one week in July 1999 of CO2 measurements made on a tall TV antenna in a forested area in Northern Wisconsin. Again, the time is recorded as UTC.
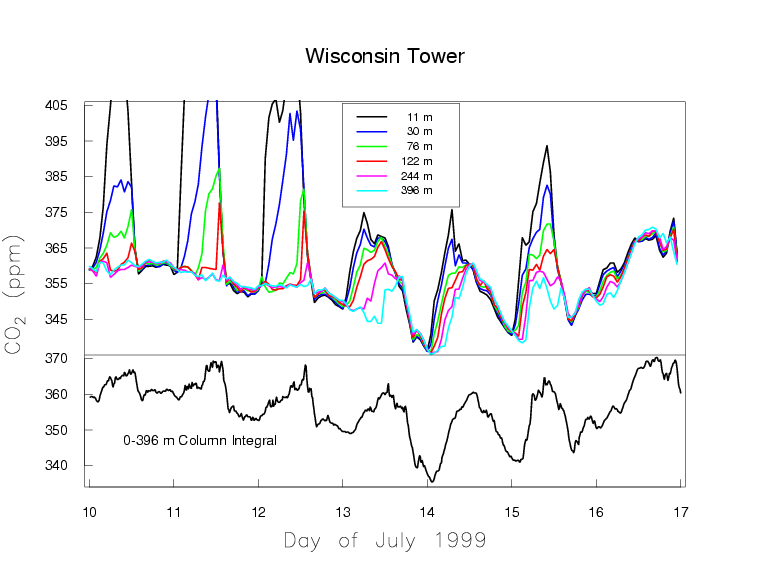 Figure 5a. Measurements of CO2 by a continuous in-situ analyzer at a tall tower during the summer in northern Wisconsin. The top plot show the CO2 values at six different heights on the tower.
Figure 5a. Measurements of CO2 by a continuous in-situ analyzer at a tall tower during the summer in northern Wisconsin. The top plot show the CO2 values at six different heights on the tower.
In Wisconsin the local time is lagging UTC by 6 hours. The measurements are made at six height levels on the tower, from 11 m to 396 m above the ground. Near the ground there is a very large diurnal cycle which decreases with altitude. At sunset, when photosynthesis shuts down, the CO2 mole fraction shoots up, especially at the lowest levels, because plants and soils keep respiring during day and night, releasing CO2. Sunset occurs at around 1 am UTC, which equals 7 pm local time of the previous day at the tower. During the day photosynthesis is stronger than respiration, causing net removal of CO2 from the atmosphere, and the CO2 gradient along the height of the tower reverses. Now the 11 m level near the ground has the lowest CO2 mole fraction. The mole fraction differences between the levels are very small during the day because the air is vigorously mixed, typically up to altitudes of 1-2 km. During the night the ground cools and the atmosphere becomes stable, with little vertical mixing because the coldest (densest) air is near the ground. The respired CO2 is trapped in the stable boundary layer near the ground, which may have a thickness of only tens of meters. The buildup of respiratory CO2 near the ground is more strongly dependent on the atmospheric stability, driven by the weather, than on the rate of respiration. It is more difficult to quantify emissions/removals of CO2 over the continent than at background sites like Mauna Loa that average over very large areas. Figure 5a also shows slow variations of CO2 on the time scale of weather systems, several days to a week, with the wind bringing air masses from different directions.
Figure 5b presents one week in January which portrays a typical situation in the winter. There are no strong sources/sinks of CO2 in the vicinity of the tower, because we see fairly uniform mole fractions along the height of the tower. The variations on the time scale of weather systems remain, due to sources/sinks far away from the tower.
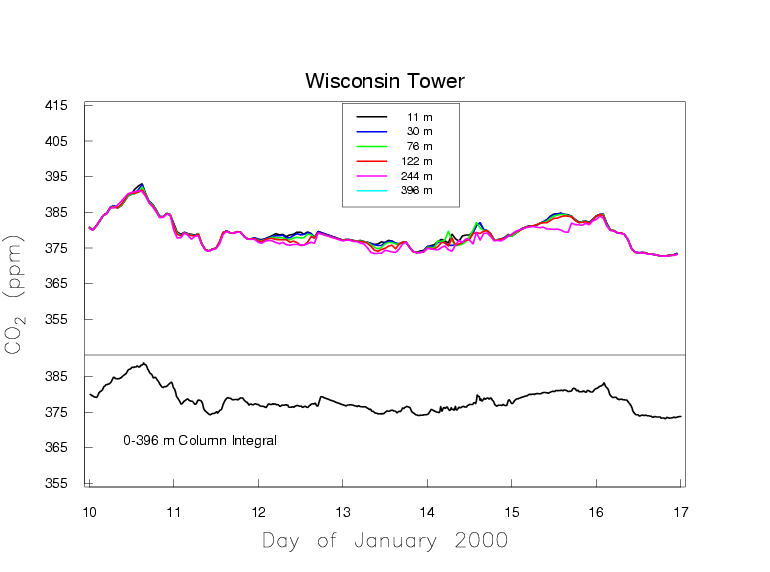 Figure 5b. Measurements of CO2 by a continuous in-situ analyzer at a tall tower during the winter in northern Wisconsin.
Figure 5b. Measurements of CO2 by a continuous in-situ analyzer at a tall tower during the winter in northern Wisconsin.